Do Kliniki Gastroenterologii i Hepatologii w Katowicach został przyjęty 67‑letni mężczyzna w celu diagnostyki choroby cholestatycznej wątroby z towarzyszącą trombocytopenią. Pięć miesięcy wcześniej pacjent został zdyskwalifikowany od zabiegu elektywnej cholecystektomii z powodu zmniejszonej liczby płytek krwi, które wahały się w granicach 50‑70 x 103/μl (wartości referencyjne: 130‑400 x 103/μl) oraz wydłużenia czasu protrombinowego do 17,4 s (wartości referencyjne: 9,4‑12,5 s). Chory skarżył się na zmniejszającą się od około 2 lat tolerancję wysiłku fizycznego oraz utratę masy ciała o 12 kg w ciągu ostatnich 2 miesięcy. Poza kamicą pęcherzyka żółciowego i nadciśnieniem tętniczym w wywiadzie nie stwierdzono innych obciążeń medycznych.
W badaniu fizykalnym uwagę zwracały powiększona wątroba i śledziona, a także zmiany na skórze tułowia i kończyn dolnych o charakterze wysypki plamisto‑grudkowej o średnicy zmian około 0,5 cm (ryc. 1).

Ryc. 1. Plamisto‑grudkowa wysypka podudzia.
W badaniach laboratoryjnych potwierdzono zaburzenia w układzie krzepnięcia, a także zwiększenie aktywności fosfatazy zasadowej do 390 U/l (wartości referencyjne: 38‑126 U/l) i γ‑glutamylotranspeptydazy do 109 U/l (wartości referencyjne < 55 U/l). Aktywności aminotransferaz, wskaźnik INR oraz stężenie bilirubiny były prawidłowe. W badaniu ultrasonograficznym (USG) jamy brzusznej stwierdzono powiększenie wątroby do 14 cm w wymiarze przednio‑tylnym, a wymiar podłużny śledziony wynosił 15 cm. Badanie endoskopowe górnego odcinka przewodu pokarmowego nie wykazało obecności zmian patologicznych. W kolonoskopii błona śluzowa jelita grubego była miejscami granulowana. Tomografia komputerowa jamy brzusznej potwierdziła powiększenie wątroby i śledziony oraz ujawniła obecność powiększonych węzłów chłonnych w przestrzeni zaotrzewnowej. W miąższu wątroby widoczne były nieliczne słabo odgraniczone obszary o obniżonej densyjności, które w pierwszej kolejności sugerowały obecność zmian przerzutowych. Ponadto w odcinku lędźwiowym kręgosłupa opisano zmiany osteosklerotyczne o wyglądzie „kręgów z kości słoniowej”.
Ze względu na niejasny charakter choroby zdecydowano o wykonaniu biopsji wątroby. Badanie histopatologiczne pobranego materiału wykazało obecność intensywnego nacieku złożonego z mastocytów. W badaniach immunohistochemicznych stwierdzono dodatni odczyn na obecność CD117, tryptazy mastocytowej oraz CD25 i ujemny dla CD1a. Również barwienia materiału pobranego w trakcie kolonoskopii wykazały w obrębie błony śluzowej jelita grubego obecność komórek tucznych z typowym dla tych komórek profilem immunohistochemicznym.
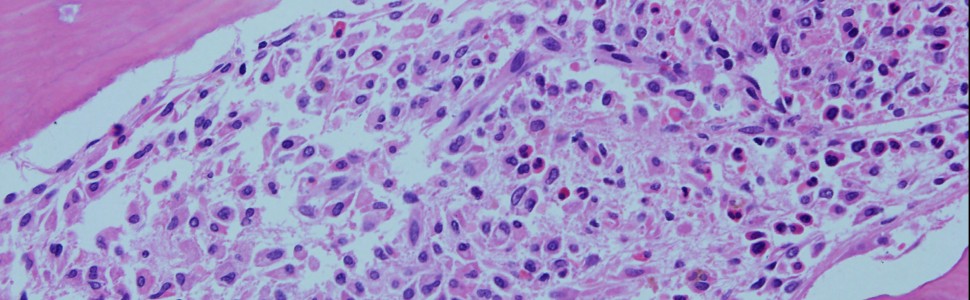
Po rozpoznaniu układowej mastocytozy chory został przekazany do Kliniki Hematologii i Transplantacji Szpiku w Katowicach. Biopsja szpiku wykazała naciek wrzecionowatych mastocytów zajmujący 50% utkania szpikowego, z typowym dla tych komórek obrazem barwienia immunohistochemicznego (ryc. 2).

Ryc. 2. Biopsja szpiku kostnego – naciek wrzecionowatych komórek odpowiadających mastocytom.
Stężenie tryptazy w osoczu wynosiło 200 ng/ml (wartości referencyjne < 11,4 ng/ml). Badania molekularne wykluczyły obecność mutacji D816V c‑kit oraz FIP1L1‑PDGFRA. U chorego rozpoczęto terapię interferonem‑α (Intron A) s.c. w dawce 3 mln j.m. 2 razy w tygodniu. Chory kontynuuje terapię od 6 miesięcy, jednak ani obraz histopatologiczny szpiku, ani stężenie osoczowe tryptazy nie wykazują istotnej poprawy.
Dyskusja
Termin „mastocytoza” odnosi się do heterogennej grupy rzadkich chorób proliferacyjnych, w przebiegu których dochodzi do namnażania się mastocytów lub ich prekursorów. Komórki te naciekają jeden narząd/jedną tkankę lub więcej, najczęściej występując w obrębie skóry i szpiku kostnego [2, 3]. Klasyfikacja Światowej Organizacji Zdrowia (WHO) dzieli mastocytozę na kilka podtypów, wyróżniając oprócz postaci skórnej postać układową o powolnym przebiegu (indolent systemic mastocytosis), mastocytozę układową z klonalnym rozrostem linii komórkowych niepochodzących z komórek tucznych, agresywną mastocytozę układową, białaczkę i chłoniaka mastocytowego oraz guza zbudowanego z komórek tucznych (mastocytoma) [4]. Obraz kliniczny i rokowanie w poszczególnych postaciach wykazują znaczne różnice, od przebiegu praktycznie bezobjawowego, niewpływającego na czas przeżycia, do form wysoce agresywnych, ze słabą odpowiedzią na leczenie (około 3% przypadków) [5, 6].
Rozpoznanie mastocytozy układowej może być trudnym zadaniem ze względu na małą znajomość tej choroby i skomplikowane kryteria diagnostyczne. „Mastocytoza skóry” (mastocytosis in the skin – MIS) jest często pierwszym objawem tej choroby i przeważnie przybiera postać mało charakterystycznych plamisto‑grudkowych wykwitów [7]. Rozpoznanie MIS jest często punktem początkowym, od którego należy rozpocząć różnicowanie izolowanej postaci skórnej z mastocytozą układową. „Dużym” kryterium diagnostycznym mastocytozy układowej są wieloogniskowe nacieki z mastocytów w szpiku kostnym i/lub innych narządach (poza skórą). Ponadto wyróżnia się cztery „małe” kryteria odnoszące się do charakterystyki komórek nowotworowych. Należą do nich:
- obecność wrzecionowatych komórek w materiale biopsyjnym szpiku kostnego (> 25%) lub materiale pochodzącym z innego narządu (poza skórą),
- obecność mutacji c‑kit (w materiale komórkowym pobranym ze szpiku, narządu litego lub krwi obwodowej),
- obecność w szpiku, narządach litych lub krwi obwodowej komórek wykazujących następujący profil immunohistochemiczny: CD117(+), CD2(+) i/lub CD25(+),
- stężenie osoczowe tryptazy przekraczające 20 ng/ml.
Do rozpoznania mastocytozy układowej niezbędne jest spełnienie przynajmniej jednego kryterium „dużego” i jednego „małego” bądź co najmniej trzech kryteriów „małych” [4]. Agresywna mastocytoza układowa (aggressive systemic mastocytosis – ASM) jest niezwykle rzadką postacią choroby, stanowiącą około 5% wszystkich przypadków mastocytozy układowej [8]. W większości przypadków ASM nie występują typowe zmiany skórne, co znacznie utrudnia postawienie rozpoznania [9]. Chorzy na ASM prezentują objawy związane z naciekaniem różnych narządów przez komórki tuczne, do których zalicza się [10]:
Osteosklerotyczne zmiany kostne występujące u naszego pacjenta są obecne tylko w około 1/3 przypadków ASM. Zaburzenia parametrów funkcji wątroby w ASM są częste, a zwiększenie aktywności fosfatazy zasadowej jest najczęściej obserwowanym odchyleniem w badaniach laboratoryjnych [11]. Badania obrazowe wątroby, takie jak tomografia komputerowa lub rezonans magnetyczny, przeważnie dostarczają wyniki mało specyficzne dla mastocytozy, w pierwszej kolejności nasuwając podejrzenie obecności zmian przerzutowych [12]. W rzadkich przypadkach obrazy cholangiografii mogą naśladować stwardniające zapalenie dróg żółciowych, co znacznie opóźnia postawienie właściwego rozpoznania [13, 14].
W ciągu ostatniej dekady u chorych z ASM stosowano różne strategie terapeutyczne w zależności od stanu ogólnego, wieku i towarzyszących chorób. Najczęściej rozważane jest leczenie interferonem‑α, kladrybiną, glikokortykosteroidami, lekami z grupy inhibitorów kinazy tyrozynowej, a także talidomidem. Sugeruje się również skuteczność leczenia celowanego z zastosowaniem przeciwciał monoklonalnych, np. gemtuzumabu [15, 16]. W rzadkich przypadkach u chorych o szczególnie złym rokowaniu podejmuje się próby allogenicznego przeszczepienia szpiku kostnego [17].
Piśmiennictwo
Źródło: Żorniak M., Hartleb M., Waluga M., Pająk J., Pilch‑Kowalczyk J., Helbig G., Kyrcz‑Krzemień S.: Mastocytoza układowa – rzadka przyczyna hepatosplenomegalii. Gastroenetrologia Praktyczna 2014, 1(22), 67-71.








